Introducción
Las gammapatías monoclonales son un grupo de trastornos que se caracterizan por la proliferación clonal del plasmocito, con la consiguiente producción de una inmunoglobulina (IgG, IgA, IgD, IgE y cadenas ligeras) de componente monoclonal. Dentro de este grupo se encuentra el mieloma múltiple, que se presenta como la proliferación clonal del plasmocito en la médula ósea >10 %, y como criterios clínicos definitorios se encuentran la hipercalcemia, afectación renal, anemia y lesiones osteolíticas 1. Esta es la más frecuente de las gammapatías monoclonales, con una incidencia anual según estadísticas norteamericanas de 7 por 100.000 hombres y mujeres por año 2.
Las amiloidosis son un conjunto heterogéneo de patologías caracterizadas por el depósito de proteína amiloide generada por el mal plegamiento de las proteínas, ocasionando alteración de la histoarquitectura en distintos tejidos y afectando de manera progresiva la función del órgano comprometido 3. Esta puede ser primaria, asociada a otros tipos de discrasias sanguíneas como el mieloma múltiple, tal como se presentó en el caso expuesto en el presente reporte, o secundaria a enfermedades inflamatorias crónicas. Presenta una incidencia anual a nivel de Estados Unidos y Reino Unido de 6-10 casos por 1.000.000 personas, siendo esta la principal causa de amiloidosis en países desarrollados 4.
Presentación del caso
Paciente masculino de 70 años de edad con antecedente patológico de síndrome mielodisplásico de bajo riesgo fue remitido a la institución por presentar cardiopatía isquémica relacionada con enfermedad coronaria severa multivasos y leucopenia más trombocitopenia. Fue evaluado conjuntamente por los servicios de cardiología y hematología. Al examen físico, no se destacó ningún hallazgo relevante para el caso.
El paciente llegó desde un centro remisor con un ecocardiograma transtorácico que mostraba un patrón sugestivo de amiloidosis cardiaca (Figura 1). Ante la sospecha diagnóstica, el servicio de cardiología solicitó un ecocardiograma transtorácico con Strain longitudinal para confirmar o descartar la sospecha de enfermedad infiltrativa. El hallazgo de Strain Longitudinal Global fue -10,4 % (Patrón de Amiloidosis), y el ventrículo derecho presentaba paredes engrosadas, con un espesor de 9 mm y aspecto en vidrio esmerilado, sugiriendo miocardiopatía infiltrativa. Como resultado, se solicitó una resonancia magnética cardiaca para evidenciar realce tardío propio de la amiloidosis cardiaca y confirmar el diagnóstico.
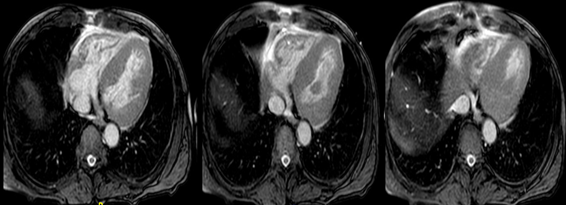
Figura 1
Amiloidosis con extenso compromiso cardiaco y con patrón de realce tardío. Fuente: archivos del caso.
Posteriormente, el paciente fue evaluado por el servicio de hematología con la sospecha de gammapatía monoclonal tipo amiloidosis cardiaca, a la espera de confirmación por resonancia magnética con patrón de realce tardío. Al mismo tiempo, se observó un deterioro de la función renal en estadio IIIB de su enfermedad renal crónica, lo cual es común en la amiloidosis sistémica. Se planteó la sospecha de estar ante un cuadro de amiloidosis sistémica, un escenario de pronóstico muy desfavorable, por lo que se solicitó una junta médica para determinar las conductas a seguir.
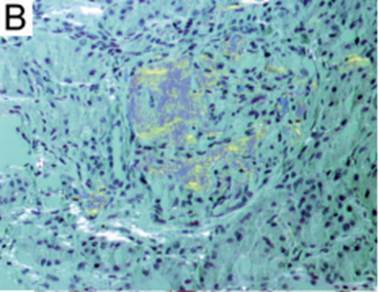
Posteriormente, se recibió el reporte oficial de resonancia magnética cardíaca, concluyendo que había amiloidosis con extenso compromiso cardiaco, con patrón de realce tardío (Figura 1), confirmando así la sospecha de amiloidosis cardiaca. Ante estos hallazgos, el servicio de hematología decidió realizar un aspirado y biopsia de hueso con tinción de rojo congo para la pesquisa de amiloidosis sistémica. Además, se solicitó una interconsulta al servicio de cirugía para la toma de biopsia de grasa abdominal y realizar la tinción de rojo congo para confirmar o descartar la presencia de amiloide con birrefringencia (Figura 2) 3.
Durante la evolución de la estancia clínica del paciente, se obtuvo la inmunotipificación por citometría de flujo en médula ósea, evidenciando plasmocitos con fenotipo neoplásico propio del mieloma múltiple. El informe reveló la deleción 17p (tp53) como negativa, translocación 4; 14 como negativa, y translocación 14; 16 también como negativa, lo que sugiere un bajo riesgo citogenético.
Posteriormente, el paciente fue llevado a quirófano para la toma de biopsia de tejido graso a nivel periumbilical. La interpretación realizada por el servicio de anatomopatología de grasa abdominal reveló depósito de material amiloide, confirmando el diagnóstico de amiloidosis sistémica asociada a mieloma múltiple.
Tras el posoperatorio de la biopsia, el paciente presentó un sangrado continuo en el sitio de la intervención que persistió más de lo esperado. Ante la imposibilidad de iniciar el tratamiento antineoplásico para la gammapatía monoclonal con fines de inducción a la remisión, el servicio de hematología decidió solicitar pruebas debido a la sospecha de alteración en la coagulación. Las pruebas incluyeron una curva de agregación plaquetaria, factor de von Willebrand y cofactor de ristocetina. Los resultados mostraron un defecto en la función plaquetaria, con ADP deficiente (38 %), colágeno ausente (6,9 %), epinefrina ausente (18 %) y ristocetina deficiente (47 %), lo que requirió la transfusión de 1 unidad de plaquetas por aféresis.
Adicionalmente, se catalogó al paciente con enfermedad de von Willebrand tipo II debido a la disminución del cofactor de ristocetina (10,0). Se repuso el déficit con factor VIII enriquecido con von Willebrand mediante tres dosis para controlar el sangrado y se dio inicio a un ciclo de poliquimioterapia buscando la inducción a la remisión. Después del último protocolo de ciclo por 21 días con lenalidomida y 9 días de descanso, se decidió realizar una intervención coronaria percutánea por el hemodinamista. Esta incluyó angioplastia de oclusión total de la coronaria derecha a nivel de la latero ventricular, ultrasonido intravascular cardíaco y angioplastia de la circunfleja distal.
Debido a la evolución intrahospitalaria favorable, se decidió dar de alta al paciente con seguimiento ambulatorio por cardiología y hematología. Se programaron próximos ciclos de quimioterapia en busca de la remisión y vigilancia periódica ambulatoria, ya que no contaba con el rendimiento clínico necesario para someterse a un trasplante de médula ósea una vez alcanzado el estado de enfermedad mínima residual.
Discusión
Las gammapatías monoclonales son un grupo de patologías que requieren un abordaje correcto a través de una historia clínica completa, teniendo en cuenta la edad y la raza, ya que estas variables influirán, sumadas a los síntomas y signos que consignemos, para pensar en su diagnóstico. Dentro de las alteraciones que involucran a la célula plasmática o plasmocito, tenemos como exponente principal al mieloma múltiple, que constituye el 65 % de los casos, representando el 1 % de las neoplasias malignas y aproximadamente el 10 % de las neoplasias hematológicas 5. En segundo lugar, se encuentra la amiloidosis, con una prevalencia del 13 %. Tanto el mieloma múltiple como la amiloidosis son más frecuentes en la población afrodescendiente, como lo era el paciente descrito en el presente reporte. Ambas enfermedades ocurren con mayor frecuencia en edades tardías, alrededor de los 60 a 70 años. Aunque el mieloma múltiple no muestra predilección por el género, cabe destacar que la amiloidosis ocurre más en hombres que en mujeres. En cuanto a la epidemiología latinoamericana, se reportó que la incidencia más alta de mieloma múltiple en países latinos se encontró en Cali (Colombia) con 14,2 casos por 100.000 habitantes 6.
En relación con la amiloidosis sistémica, es importante demostrar la presencia del depósito de proteína amiloide. Generalmente, se tiende a tomar muestras poco invasivas, como en el caso de nuestro paciente, donde se demostró la presencia de amiloide a nivel de tejido adiposo mediante biopsia de grasa abdominal. Adicionalmente, se evidenció infiltración cardiaca por datos objetivos, tanto en ecocardiograma como en resonancia magnética cardíaca. Sin embargo, cabe destacar que no contamos con la medición de la proteína Transtirretina Gold estándar para el diagnóstico 4. Es importante tener en cuenta que la mortalidad es alta en este grupo de pacientes, sobre todo por compromiso a nivel cardiaco, con el desarrollo de insuficiencia cardiaca y arritmias, como se descubrió posteriormente en la evolución del paciente, por síntomas sincopales que llevaron a la realización de Holter de 24 horas con detección de periodos de bradicardia extrema, alternando con taquicardia ventricular, lo que requirió la colocación de un cardio-desfibrilador.
Finalmente, la enfermedad de von Willebrand adquirida es poco común, probablemente porque es uno de los trastornos de la hemostasia más subdiagnosticados. Esta debe sospecharse en pacientes con aparición tardía de diátesis hemorrágica, sin antecedentes de sangrado. Tiene una fuerte asociación con enfermedades linfoproliferativas (48 %), mieloproliferativas (15 %), neoplasias (5 %), inmunológicas (2 %), cardiovasculares (21 %) y misceláneas (9 %). Generalmente, su diagnóstico ocurre simultáneamente con el diagnóstico de la enfermedad hemoproliferativa clonal asociada, en este caso, mieloma múltiple y amiloidosis sistémica 7.
Se puede concluir que debemos tener en cuenta, al momento del abordaje de un paciente con sospecha de gammapatía monoclonal, el contexto clínico y epidemiológico para acortar los tiempos en el diagnóstico y, por lo tanto, un manejo precoz que impacte en la mortalidad. Cabe resaltar dentro de las fortalezas del presente reporte que se realizó un enfoque multidisciplinario por parte de las especialidades involucradas, contando con diferentes estudios tanto imagenológicos como inmunohistoquímicos de una complejidad avanzada, los cuales no se encuentran en la mayoría de los centros de atención de menor nivel de complejidad. Entre las limitaciones a mencionar, resalta el hecho de no contar con la prueba de la medición de la proteína transtirretina, gold estándar para el diagnóstico de amiloidosis sistémica, ya que, a pesar de no ser tan frecuente, podemos tener casos como el del paciente descrito en el siguiente reporte, donde ocurre la asociación del mieloma múltiple y la amiloidosis sistémica en el contexto de una enfermedad de von Willebrand.