Los trastornos neurocognitivos y retos de la epigenética
Neurocognitive Disorders and Challenges of Epigenetics
Recibido: 20/06/2022
Aceptado: 28/09/2022
Autores
Juan David Marenco Salazar
José María Mejía Barrera
Javier Alfredo Pérez Martínez
Said Samir Sánchez Salcedo
Alfredo Pugliese Jiménez
Resumen
Introducción: los trastornos neurocognitivos se definen como un conjunto de anomalías en la capacidad de aprendizaje y memoria de un individuo. En múltiples estudios, se han encontrado diversos marcadores genéticos que emergen a raíz de la evidencia de que distintos factores ambientales tienen la capacidad de influir en la expresión de dichos marcadores. El objetivo es recopilar información actual acerca de las interacciones entre enfermedades neurológicas y la epigenética en el desarrollo de los trastornos neurocognitivos.
Metodología: Se usó la guía PRISMA para revisiones sistemáticas sobre los aspectos genéticos y epigenéticos que participan en la función sináptica y la memoria.
Resultados: los mecanismos epigenéticos desempeñan un papel crucial en el control de los procesos de modificación sináptica, en la formación y desarrollo de las funciones cognitivas. Alteraciones en estos mecanismos producen déficit cognitivo y de memoria en padecimientos neurodegenerativos.
Conclusión: Los resultados obtenidos en diferentes modelos muestran un escenario prometedor con tratamientos potenciales para algunos padecimientos.
Abstract
Introduction: neurocognitive Disorders can be defined as abnormalities in the capacity of learning and memory of an individual. In recent years, multiple predisposing genetic markers have been found that are emerging now, as a result of the evidence that different environmental factors have the ability to influence genes. To collect current information about the interactions between neurological diseases and epigenetics in neurocognitive development.
Methodology: the PRISMA guideline for systematic reviews was used on the genetic and epigenetic aspects that participate in synaptic function and memory.
Results: epigenetic mechanisms play a crucial role in synaptic modification processes and in the formation and development of memory. Alterations in these mechanisms produce cognitive and memory deficits in neurodegenerative disorders.
Conclusion: the results obtained in different models show a promising scenario with potential treatments for some conditions.
Introduction
Nyberg et al. (1), definen los trastornos neurocognitivos como principalmente anomalías en la capacidad de aprendizaje y memoria de un individuo. Esta pérdida cognitiva se adquiere durante el curso de diferentes enfermedades y, por lo tanto, representa una disminución del nivel de funcionamiento alcanzado previamente.
En una revisión sistemática realizada por Rosales et al. (2), encontraron que el estudio progresivo y cada vez más detallado del genoma humano, junto con los avances tecnológicos propios del nuevo siglo, han abierto una amplia gama de posibilidades y métodos para identificar procesos epigenéticos implícitos (modificación de las histonas, metilación del ADN, cambios en los reguladores del ARN), que tienen un impacto importante en las enfermedades neurocognitivas y que explicarían gran parte de la causalidad de estos trastornos. Así, hoy en día, se acepta que los diferentes mecanismos epigenéticos juegan un papel fundamental en los procesos de modificación sináptica, en la formación y desarrollo de la memoria en condiciones normales.
Con esta revisión pretendemos describir las diferentes modificaciones epigenéticas en estos trastornos y las posibilidades que este conocimiento supone en el desarrollo de potenciales tratamientos, lo cual a futuro podría repercutir favorablemente en la salud de nuestros pacientes.
Mecanismos epigenéticos
La epigenética es un término acuñado por Waddington para referirse al conjunto de procesos de regulación de la expresión génica que no incurren en cambios en la secuencia de nucleótidos del ADN y que tienen un carácter heredable (1). Hasta la fecha, se han descrito tres mecanismos que participan de forma importante en la regulación génica: 1) la modificación de histonas, 2) la metilación del ADN y 3) los Ácidos Ribonucleicos (ARN) no codificantes. Entre estos, la modificación de histonas es el proceso más ampliamente conocido y relacionado con un importante número de enfermedades neurológicas (2).
1. Modificación de histonas
Malecová et al. (3), explican que el genoma diploide contenido dentro de una sola célula abarca 2 metros de ADN. Esto requiere varios niveles de compactación organizada que permiten que el núcleo envuelva el ADN y que la maquinaria transcripcional pueda acceder a los genes. Esto se logra mediante la cromatina, una estructura compleja que consta de ADN y proteínas estrechamente opuestas conocidas como histonas. Cuatro proteínas centrales (H2A, H2B, H3 y H4) componen el complejo octámero de histonas. Junto con los 147 pares de bases del ADN circundante y el ADN enlazador, esto constituye el nucleosoma, la unidad fundamental de la cromatina.
Saxena et al. (4), describen que las histonas son proteínas básicas que participan en el empaquetamiento del ADN y la formación de los nucleosomas. Un trabajo realizado por Kouzarides T. revisó la acetilación de histonas provocada por las Histonas Acetiltransferasas (HAT), que activa la transcripción mediante la formación de la estructura de eucromatina, mientras que la Histona Desacetilasa (HDAC) provoca la desacetilación, lo que lleva a la represión de la transcripción mediante la formación de la estructura de heterocromatina. Ambas enzimas son responsables de la plasticidad sináptica y el procesamiento de la memoria (5).
En conjunto, Strahl et al. (6), Jiang et al. (7) y Shukla et al. (8), demostraron que la modificación de las histonas es un mecanismo que ocurre de forma independiente o como consecuencia de la metilación del ADN. La hipótesis actual indica que los extremos amino-terminales de las histonas, que sobresalen de la estructura de la cromatina, participan en un proceso de integración de señales que dan lugar a un patrón específico de modificaciones postraduccionales y forman un “código de histonas”, el cual dirige la actividad de numerosos factores y cofactores de la maquinaria transcripcional. Actualmente se conocen 4 tipos de modificaciones postraduccionales en los extremos de las proteínas histónicas que participan en el marcaje epigenético: 1) acetilación, 2) metilación, 3) ubiquitinación y 4) fosforilación.
Salozhin et al. (9), encontraron que la acetilación de histonas en la corteza insular provoca la formación de recuerdos. Se informa que los inhibidores de HDAC aumentan la magnitud de las sinapsis de CA1. Por lo tanto, se ha planteado la hipótesis de que la mejora de la memoria podría ser posible mediante inhibiciones de HDAC. Existen medicamentos como Tricostatina A (TSA), Ácido Valproico (VPA), el Ácido Suberoilsanilidohidroxámico (SAHA), nicotinamida y el fenilbutirato de sodio (4-PBA) que inhiben las enzimas HDAC (Figura 1).
2. Metilación del ADN:
Feng et al. (10), corroboraron que la metilación del ADN es responsable del silenciamiento de la expresión génica. Esta metilación del ADN es un proceso epigenético que participa en la regulación de la expresión génica de dos maneras: directamente al impedir la unión de factores de transcripción, e indirectamente al propiciar la estructura “cerrada” de la cromatina. El ADN se presenta como regiones de 1.000-1.500 pares de bases ricas en dinucleótidos citosina-fosfato-guanina (islas CpG), que son reconocidas por las enzimas Histonas Metiltransferasas (HMT), las cuales metilan el ADN durante su replicación, manteniendo así la memoria del estado metilado en la molécula hija de ADN. En general, esta modificación se vuelve estable y se hereda como un patrón clonal de metilación. Por otro lado, en condiciones normales, las enzimas histonas desmetilasas (HDM) desmetilan el ADN, permitiendo su correcta transcripción. La sobremetilación del ADN mantiene la estructura condensada de la cromatina e impide la transcripción de los genes involucrados. También existen otros medicamentos que intervienen en la metilación del ADN, como 5-aza-2-deoxicitidina (5-aza), decitabina, bisulfito y zebularina, los cuales inhiben las enzimas HMT.
Xicota et al. (11), encontraron que las metiltransferasas de ADN (DNMT) como DNMT1, DNMT3A y DNMT3B también causan metilación en el ADN. En uno de los estudios, se ha demostrado que cuando se eliminan DNMT1 y DNMT3A en un ratón, se encuentra una pérdida del aprendizaje y la memoria basados en el hipocampo. Un inhibidor de DNMT llamado epigalocatequina-3-galato (EGCG), el principal polifenol del té verde (Camellia sinensis), ha ganado especial interés como posible tratamiento después de que estudios observacionales relacionaron el consumo de té verde con un menor riesgo de demencia.
3. ARN no codificantes
En los trabajos de Tang et al. (12), se describió cómo existen cadenas de ARN que no codifican para ninguna proteína, pero cuyas secuencias son complementarias al ADN o ARN codificante, y esto impide su traducción. Esta es una forma de regulación negativa de la expresión a nivel postranscripcional. A este tipo pertenecen los denominados ARN de interferencia (iARN), los cuales se unen a secuencias complementarias de ARNm para degradarlos e impedir su traducción en proteínas.
Los ARN de interferencia son moléculas pequeñas, de 20 a 25 nucleótidos, que se generan por fragmentación de precursores más largos. Se pueden clasificar en tres grandes grupos de moléculas: siARN (del inglés small interfering RNA, en español, ARN interferente pequeño), miARN (en inglés, micro-ARN o miARN), y piARN (en inglés, Piwi-interacting RNAs, ARN asociados a Piwi) (13). De estos ARN de interferencia, los micro-ARN son una diana terapéutica potencial para la regeneración neuronal (12).
Figura 1. Trastornos neurocognitivos y alteraciones epigenéticas
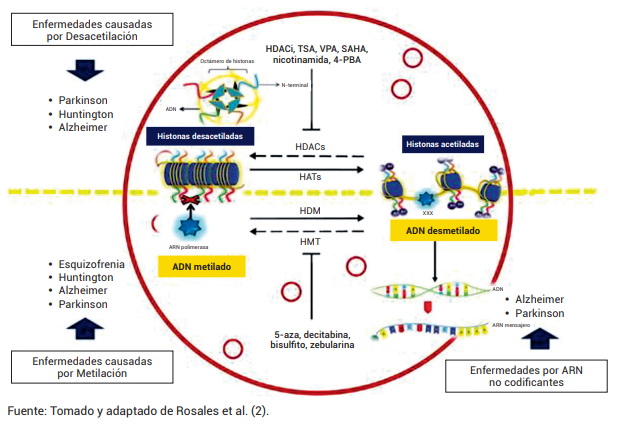
Trastornos neurocognitivos y alteraciones epigenéticas
1. Enfermedad de Alzheimer
La World Health Organization (WHO) (14), define la Enfermedad de Alzheimer (EA) como un trastorno neurodegenerativo crónico dependiente de la edad que se caracteriza clínicamente por el deterioro progresivo de la memoria y las funciones cognitivas. Es la causa más frecuente de demencia degenerativa primaria en todo el mundo, afectando a alrededor de 50 millones de personas y con casi 10 millones de casos nuevos cada año.
Los trabajos de Masters (15), explican cómo la acumulación de placas de amiloide-β (Aβ) y ovillos neurofibrilares en diferentes regiones del cerebro son las características más importantes de la EA. La proteína precursora amiloide (APP) y los genes de presenilina 1 o 2, si se mutan, causan una EA autosómica dominante. Igualmente, Guan et al. (16), explicaron cómo la generación de péptidos Aβ patógenos requiere de β-secretasa (BACE1), que escinde la proteína precursora amiloide (APP); el paso limitante de la velocidad en la producción de Aβ. Evidencia reciente demuestra que la acetilación de histonas y la metilación del ADN participan en la etiología de la EA
Guan et al (16), también describieron que las HDAC de clase 1 (HDAC2 y HDAC3) se expresan en una mayor concentración en la región del cerebro asociada con la memoria. Se observó un aumento en la fagocitosis de amiloide microglial en un modelo de ratón con EA cuando se suprimieron los genes de HDAC de clase 1. HDAC2 disminuye la acetilación de histonas de genes responsables de la memoria y el aprendizaje. En un estudio de la EA, se informó sobre la sobreexpresión de HDAC2 en la corteza prefrontal y el hipocampo en un modelo de ratón. Estos y estudios anteriores indican que la modificación de histonas juega un papel vital en el desarrollo de la EA (17).
Saura et al. (18), explican cómo la metilación del ADN es otro mecanismo implicado en la etiología de la EA, siendo el proceso de hipometilación el más ampliamente reportado. En cultivos celulares, la hipometilación de la región promotora del gen PS1 incrementó la expresión de presenilina y potenció la formación de las placas β-amiloide. Igualmente, Song C. determinó que la metilación de 5mC en el ADN tiene un papel importante en la expresión de genes neuronales y el desarrollo neuronal. Estos productos desempeñan un papel fundamental en el desarrollo y la función de las neuronas. En los pacientes con EA, se ha observado que las firmas de 5mC, 5Fc, 5-hidroximetilcitosina (5hmC) y 5caC son limitadas, lo que provoca la degeneración de las neuronas. Las citosinas en la región 207-182 de APP están metiladas en el cerebro normal, pero con el envejecimiento, estas citosinas se desmetilan y conducen a la deposición de Aβ en el cerebro. Se encontró que, en el hipocampo, el cerebelo y la corteza entorrinal de los pacientes con EA, se redujeron los niveles de 5mC (19).
2. Enfermedad de Parkinson
Se trata de la segunda enfermedad neurodegenerativa más frecuente después de la EA. En el año 2016, se estimó que 6,1 millones de personas en el mundo sufrían la enfermedad, lo que representó un aumento de más del doble en comparación con la cifra de 1990 (2,5 millones). En el 2040 se estima que habrá alrededor de 17 millones de afectados. Para Poewe et al. (20), esto hace que la enfermedad de Parkinson (EP), sea de todas las enfermedades neurológicas, la de más rápido crecimiento a nivel mundial según el estudio de carga global de la enfermedad.
Desplats et al. (21), describieron cómo la EP es causada por la muerte de neuronas dopaminérgicas que contienen neuromelanina en la sustancia negra del cerebro, con presencia de cuerpos de Lewy. La EP es el resultado de una combinación de herencia poligénica, exposición ambiental y también una compleja interacción gen-ambiente. Los genes regulados epigenéticamente que participan en la EP son LRRK2, Parkin, PARK16/1q32 y GPNMB, además de la α-sinucleína (SNCA).
Cuando Goers et al. (22), realizaron un análisis epigenético en el cerebro de la EP, se reveló que el gen SNCA podría estar regulado epigenéticamente. Se ha encontrado hipermetilación en SNCA en individuos con alcoholismo y anorexia, lo que da evidencia de regulación epigenética de SNCA. Se observó reducción de la metilación del ADN en el intrón 1 de SNCA en varias regiones del cerebro de pacientes con EP esporádica. Se observa un mayor nivel de expresión de SNCA si se inhibe la metilación del ADN. Como sabemos, DNMT1 es responsable de la desmetilación, aumentando así la concentración de α-sinucleína en el cerebro. Además, se ha demostrado que SNCA secuestra DNMT1 y, en consecuencia, conduce a alteraciones epigenéticas en el cuerpo de Lewy. A diferencia de SNCA, la metilación del promotor del gen de Parkin no causa ningún efecto en el desarrollo o patogénesis de la EP (21).
Por otro lado, estudios demostraron que la inhibición de la acetilación de histonas ocurre cuando la α-sinucleína se une a la histona, por lo que los inhibidores de HDAC protegen a la neurona contra la neurotoxicidad mediada por la α-sinucleína (22). Sharma et al. (23), igualmente encontraron que la regulación de la maduración y función de las neuronas dopaminérgicas en el mesencéfalo se realiza mediante el microARN miR-133b. En el mesencéfalo de pacientes con EP se ha observado deficiencia de miR-133b. Estudios recientes han demostrado que los miARN están implicados en la EP (24).
Tarale et al. (25), también describieron cómo la pirosecuenciación de la región del cerebro mostró diferencias en los niveles de metilación del ADN en la sustancia negra en las islas CpG. La secuenciación de bisulfito es el método más reciente y el “estándar de oro” para descubrir la metilación del ADN. Se ha observado que factores ambientales y nutricionales como el manganeso (Mn), el café, los pesticidas, la deficiencia de folato y algún polimorfismo de un solo nucleótido juegan un papel vital en la metilación del ADN. Un estudio con células SHAY5Y (neuroblastoma humano dopaminérgico) mostró una alteración en la función de PARK2 y PINK1 debido a la hipermetilación del ADN tras la exposición a Mn. La hipermetilación en SNCA, NPAS2, los genes PGC1-α y CYP2E1 también son responsables de la EP.
3. Enfermedad de Huntington
La Enfermedad de Huntington (EH) es un trastorno neurodegenerativo hereditario autosómico dominante causado por una expansión de repetición de trinucleótidos CAG en el exón 1 del gen de la huntingtina (HTT). Es considerada una enfermedad devastadora que afecta a aproximadamente 1-2 de cada 10.000 personas en el mundo y se acompaña de disfunción neuronal y neurodegeneración. La EH se manifiesta como un corea progresivo, con disminución de las capacidades mentales, acompañada de problemas conductuales, emocionales y psiquiátricos, que progresan a demencia y, en última instancia, a la muerte (26).
Alcalá et al. (27), encontraron que el número de repeticiones de trinucleótidos CAG en HTT es polimórfico en la población normal y no supera los 36-39 CAG. Por encima de este umbral, la repetición CAG es patógena, lo que da como resultado la producción de proteínas HTT mutadas tóxicas con una expansión de poliglutamina (polyQ) muy propensa a la agregación. A pesar de la expresión ubicua del gen HTT, polyQ-HTT es predominantemente tóxico para las neuronas específicas del cuerpo estriado. Sin embargo, a medida que avanza la enfermedad, se ven afectadas regiones cerebrales adicionales, incluidas las regiones corticales. La microglía y los astrocitos también contribuyen al proceso patogénico.
Hoy en día, la EH es una de las enfermedades neurodegenerativas más estudiadas en el contexto de la regulación epigenética. La neurodegeneración es el resultado de un largo período de disfunción de las células cerebrales, en donde la mutación de la EH induce desregulaciones transcripcionales y epigenéticas a gran escala en el cerebro de estos pacientes.
Los modelos de EH y el tejido cerebral humano con EH muestran alteraciones en la expresión génica, un aumento de los dominios de heterocromatina H3K9me3 y, lo que es más importante, la histona acetiltransferasa CBP está mal localizada en los agregados de poliglutamina en células cultivadas con EH, en modelos de ratón y en cerebros post mortem. Diversos estudios de modelos de EH identificaron inicialmente un efecto beneficioso de los inhibidores de HDAC para mitigar drásticamente la neurodegeneración y sugirieron que la disminución de la acetilación de histonas podría estar involucrada en los mecanismos de la EH. Es sorprendente que los inhibidores de HDAC puedan proteger contra la neurodegeneración tanto en los modelos de EH como en los de EA, a pesar de los diferentes mecanismos patogénicos subyacentes.
Los esfuerzos recientes realizados por Seredenina et al. (28), para atacar la metilación de histonas en la EH muestran que la reducción de los niveles de H3K9me3 con el fármaco remodelador de la cromatina Nogalamicina ralentiza la progresión de la enfermedad en ratones transgénicos con EH, que expresan el exón 1 de HTT con 150 repeticiones de CAG. Otro mecanismo epigenético descrito por Francelle et al. (29), en la EH está relacionado con la acetilación y metilación de las histonas. Los modelos murinos en los cuales la actividad de la proteína de unión a CREB (CREBBP) está comprometida exhiben una reducción en la acetilación de la cromatina, disminución de la plasticidad en el hipocampo y déficit en la memoria a largo plazo.
También se ha observado que la pérdida de la función de CREBBP se asocia con la degeneración del cuerpo estriado en modelos murinos de la EH. Además, la huntingtina resultante de la expresión del gen HTT mutado interactúa directamente con el dominio de las acetiltransferasas CREBBP, lo que resulta en una reducción de la actividad de esta enzima (2).
4. Esquizofrenia
La esquizofrenia se considera un trastorno neuropsiquiátrico caracterizado por alucinaciones y delirios, aislamiento social, habla desorganizada y otros síntomas, por lo general sin evidencia de patología cerebral estructural clara. Los estudios epidemiológicos sobre la esquizofrenia han encontrado tasas de incidencia de entre 7,7 y 43 casos nuevos por cada 100.000 habitantes. La prevalencia puntual es aproximadamente 5 por 1000 habitantes y el riesgo de morbilidad a lo largo de la vida está en torno al 1 %. La tasa de mortalidad es 2,5 veces mayor que la correspondiente a la población general, y los hombres tienen un riesgo de 30-40 % mayor que las mujeres, y una edad de aparición mucho más temprana (30).
La esquizofrenia es otra de las patologías en las que se han descrito mecanismos epigenéticos para comprender la fisiopatogenia y el riesgo de desarrollar la enfermedad. Existe evidencia de que la transcripción genética desregulada, indicativa de un circuito neuronal comprometido, contribuye a la función cerebral desordenada en la psicosis y los trastornos del espectro del estado de ánimo. En los trabajos realizados por Freudenreich et al. (31), aunque ninguna transcripción de un solo gen se ve afectada de manera constante, las alteraciones en los niveles de ARN contribuyen a defectos en la neurotransmisión inhibitoria GABAérgica y, de manera más general, en la organización y función de las sinapsis, el metabolismo y las funciones mitocondriales y la patología de los oligodendrocitos.
Los cambios en la acetilación de los residuos de histona lisina, que están ampliamente asociados con la expresión génica activa, impactan los patrones de expresión genética en el cerebro y, por lo tanto, influyen en las funciones emocionales y cognitivas. Estudios realizados por Cyril et al. (32), en ratones expuestos a tratamiento sistémico o inyecciones intracraneales de inhibidores de histona desacetilasa de clase I/II (HDACi) exhiben cambios de comportamiento que recuerdan a los provocados por fármacos antidepresivos convencionales. Asimismo, fármacos como el ácido valproico inducen la hiperacetilación de histonas cerebrales en un grupo selecto de promotores de genes cuando se administra a animales en dosis comparativamente altas. Por el contrario, la sobreexpresión de HDAC seleccionadas en estructuras neuronales implicadas en la neurobiología de la depresión, incluido el hipocampo, desencadena un fenotipo conductual pro-depresor. Por lo tanto, el equilibrio ordenado entre las actividades de histona acetiltransferasa y desacetilasa es crucial para el rendimiento cognitivo y la plasticidad sináptica y conductual.
Por otra parte, se considera que las perturbaciones de la metilación del ADN en la psicosis grave también pueden deberse a la actividad anormal de las DNMT o cambios en los niveles de donantes y cofactores de grupos metilo que afectan la metilación del ADN. Se ha informado que los genes DNMT están regulados positivamente en la corteza prefrontal de pacientes con esquizofrenia.
Por último, los cambios en la expresión del gen de novo ADN metiltransferasa DNMT3a en el sistema de recompensa cerebral de ratones adultos regulan comportamientos adictivos y fenotipos similares a los depresivos. De manera similar, se ha demostrado que varios medicamentos psicotrópicos producen cambios epigenéticos en el cerebro, aunque todavía no está claro si los efectos terapéuticos son una consecuencia directa de estas alteraciones epigenéticas (33).
Conclusión
Los mecanismos epigenéticos se han establecido como reguladores necesarios de la fisiología sináptica y el desarrollo de la memoria. Hasta la fecha, se conoce que desempeñan un papel central en las funciones cerebrales, por lo que cualquier alteración puede conducir a trastornos en el desarrollo neurológico o a procesos neurodegenerativos. Los diversos estudios de los que disponemos hasta el momento han generado la mayor parte del conocimiento y nos muestran un escenario promisorio con una serie de tratamientos potenciales, como los inhibidores de las HDAC, en la terapia de algunos padecimientos neurológicos. Sin embargo, a pesar de la creciente evidencia científica relacionada, aún existen una cantidad de interrogantes no resueltos con respecto a estos mecanismos y su control sobre el desarrollo o afectación de la memoria a largo plazo.
Declaración de conflicto de intereses
Los autores declaran no tener ningún conflicto de interés.
Referencias
- 1. Nyberg L., Boraxbekk C., Sorman D., Hansson P., Herlitz A., Kauppi K., et al. Biological and environmental predictors of heterogeneity in neurocognitive ageing. Ageing Research Reviews. 2020;64:1-23. https://doi.org/10.1016/j.arr.2020.101184.
- 2. Rosales M., Ochoa A., Juárez C., Barros P. Mecanismos epigenéticos en el desarrollo de la memoria y su implicación en algunas enfermedades neurológicas. Neurología. 2016;31(9):628-638. doi:10.1016/j.nrl.2014.02.004.
- 3. Malecová B., Morris KV. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. CurrOpin-MolTher. 2010;12:214-225.
- 4. Saxena A., Carninci P., Long non-coding RNA modifies chroma-tin: Epigenetic silencing by long non-coding RNAs. Bioessays.2011;33:830. doi: 10.1002/bies.201100084.
- 5. Kouzarides T., Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005.
- 6. Strahl B., Allis C. The language of covalent histone modifications. Nature. 2000;403:41-45. doi: 10.1038/47412.
- 7. Jiang Y., Langley B., Lubin F., Renthal W., Wood M., Yasui D., et al. Epigenetics in the nervous system. J Neurosci.2008;11:753-759. doi: 10.1523/JNEUROSCI.3797-08.2008.
- 8. Shukla S., and Tekwani B. Histone Deacetylases Inhibitors in Neurodegenerative Diseases. Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020. https://doi.org/10.3389/fphar.2020.00537).
- 9. Salozhin SV., Prokhorchuk EB., Georgiev GP. Methylation of DNA of the Major Epigenetic Markers. Boichemistry. 2005;70:525–532. doi: 10.1007/s10541-005-0146-8.
- 10. Feng J., Zhou Y., Campbell SL., Le T., Li E., Sweatt DJ. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010:423-430. doi:10.1038/nn.2514.
- 11. Xicota L, and De la Torre R. Epigallocatechin-3-gallate and Alzheimer’s disease. En The Neuroscience of Dementia. Martin C and Preedy V (Editors). UK. 1st Edition. 2020; Vol 2. eBook ISBN:9780128160442.
- 12. Tang X. and Sun C. The roles of MicroRNAs in neural regenerative medicine. Experimental Neurology. 2020;332:1133-94. doi: 10.1016/j.expneurol.2020.113394
- 13. Levenson JM., Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108-18.9. doi: 10.1038/nrn1604.
- 14. World Health Organization (WHO). Dementia. http://www.who.int/mediacentre/factsheets/fs362/en/ (2017).
- 15. Masters C. Alzheimer’s disease. Nat. Rev. Dis. 2015;1:150-156. doi: 10.1038/nrdp.2015.56.
- 16. Kandalepas P. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126:329-352. doi: 10.1007/s00401-013-1152-3.
- 17. Guan JS, Haggarty S, Giacometti E, Dannenberg JH, Joseph N, Gao J et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009:55-60. doi: 10.1038/nature07925.
- 18. Saura C., Choi S., Beglopoulos V., Malkani S., Zhang D., karanarayana R., et al. Loss of presenilin function causesimpairments of memory and synaptic plasticity followed by age-dependent neurodegenaration. Neuron. 2004;42:23-36. doi: 10.1016/s0896-6273(04)00182-5.
- 19. Song C., Szulwach K. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68-72. doi: 10.1038/nbt.1732.
- 20. Poewe W., Seppi K., Tanner C., Halliday G., Brundin P., Volkmann J., et al. Parkinson disease. Nat Rev Neurol. 2017;3(6):1-21. doi: 10.1038/nrdp.2017.13.
- 21. Desplats P. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J BiolChem. 2011;286:9031-9037.
- 22. Goers J., Manning A. Di Monte Nuclear localization of α-synuclein and its interaction with histones. Biochemistry. 2003;42:8465-8471. doi: 10.1021/bi0341152.
- 23. Sharma S., and Taliyan R. Targeting histone deacetylases: a novel approach in Parkinson’s disease. Parkinsons Dis. 2015:3032-94. https://doi.org/10.1155/2015/303294.
- 24. Khoo SK. Plasma-based circulating microRNA biomarkers for Parkinson’s disease. J. Parkinsons. 2012;2:321-331. doi: 10.3233/JPD-012144.
- 25. Tarale P., Sivanesan S., Daiwile A., Stoger R., Naoghare P., Pramar D. Global DNA methylation profiling of manganese-exposedhuman neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch Toxicol.2017;91:2629-41. doi: 10.1007/s00204-016-1899-0.
- 26. Narayana P., Reida S., Scottera E., McGregord A., Mehrabia N., Singh-Bainsa M., et al. Inconsistencies in histone acetylation patterns among different HD model systems and HD post-mortem brains. Neurobiology of Disease. 2020;146:1050-1092. doi: 10.1016/j.nbd.2020.105092.
- 27. Alcala-Vida R, Awada A, Boutillier A, Merienne K. Epigenetic mechanisms underlying enhancer modulation of neuronal identity, neuronal activity and neurodegeneration. Neurobiology of Disease.2021;147:1051-1055. doi: 10.1016/j.nbd.2020.105155.
- 28. Seredenina T, Luthi-Carter R. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol Dis. 2012; 45: 83–98. doi: 10.1016/j.nbd.2011.07.001.
- 29. Francelle L., Lotz C., Outeiro T., Brouillet E., Merienne K. Contribution of neuroepigenetics to Huntington’s disease. Front Hum Neurosci. 2017;11. https://doi.org/10.3389/fnhum.2017.00017.
- 30. Gavin D., Akbarian S. Epigenetic and post-transcriptional dysregulation of gene expression in schizophrenia and related disease. Neurobiology of Disease. 2012;46:255–262. doi: 10.1016/j.nbd.2011.12.008.
- 31. Freudenreich O., Brown H., Holt D. Psicosis y esquizofrenia. Massachusetts General Hospital. Tratado de Psiquiatría Clínica. Elsevier, España. 2018;28:307-323.
- 32. Cyril P., Akbarian S. Equilibrio de las actividades de metilación de histonas en trastornos psiquiátricos. Trends in Molecular Medicine. 2011;17(7):372-379. doi: 10.1016/j.molmed.2011.02.003.
- 33. Covington H. Un papel de la metilación represiva de histonas en la vulnerabilidad al estrés inducida por la cocaína. Neuron. 2011;71:656-670.