Introducción
La deficiencia de α1-Antitripsina (DAAT) es un trastorno genético poco distinguido. Su ausencia no se caracteriza por ser una entidad patológica específica, pero se asocia con la predisposición a la génesis de numerosos trastornos pulmonares y hepáticos, principalmente el enfisema pulmonar 1. Los portadores de deficiencia severa tienen una esperanza de vida más baja que la población general y un gran impacto en su calidad de vida, relacionado con las complicaciones de las diferentes patologías 2.
La α1-Antitripsina (AAT) es una glicoproteína compuesta por una cadena de 394 aminoácidos y tres cadenas laterales de carbohidratos, codificada por el gen SERPINA1, ubicado en el brazo largo del cromosoma 14 (14q32.1) 2. Pertenece a las familias de las serpinas, es producida por los hepatocitos y actúa como un inhibidor de la serina proteasa circulante cuyo sustrato principal es la elastasa de neutrófilos 2,3.
El DAAT es de herencia autosómica codominante mediante dos alelos, uno de cada progenitor 3,4. Este trastorno genético se caracteriza por la destrucción proteolítica del tejido pulmonar, dependiente en gran medida de la elastasa de los neutrófilos 3. Además, los polímeros de AAT retenidos en el retículo endoplásmico de los hepatocitos pueden causar daño hepático con una presentación clínica variable, desde hepatitis neonatal hasta cirrosis hepática y carcinoma hepatocelular en adultos, mientras que la falta de proteína circulante puede promover el desarrollo de la Enfermedad Pulmonar Obstructiva Crónica (EPOC) 3,5.
La gravedad de los síntomas es muy variable y habitualmente se manifiestan por primera vez en la cuarta o quinta década de la vida 5,6. En estudios de función e imágenes pulmonares, aunque es más característico el enfisema panlobulillar, también es común encontrar un patrón de enfisema centrolobulillar 3,5,7.
La deficiencia es posible identificarla mediante la cuantificación de niveles séricos de la proteína y posteriormente el genotipado del locus del Inhibidor de la Proteasa (IP), con el uso de tecnología de reacción en cadena de la polimerasa (PCR) o polimorfismos de longitud de fragmentos de restricción (RFLP) 8.
Estas pruebas pueden detectar el alelo M normal y las variantes patogénicas conocidas más comunes (S y Z), expresando fenotipos de proteínas MM, MS, MZ, SS, SZ y ZZ que representan más del 95 % de todas las variantes conocidas, especialmente PI*SZ (que expresa aproximadamente el 50-60 % de AAT) y PI*ZZ (que expresa aproximadamente el 10-20 % de AAT) 6,8,9. La DAAT grave está definida por una concentración sérica inferior al 35 % del valor medio esperado o 50 mg/dL por nefelometría 10.
En la actualidad, la infusión de ATT purificada es el tratamiento para la DAAT, aunque solo con evidencia en pacientes con enfisema y genotipos PI*ZZ, PI*SZ, PI*Z-nulo, Pinulo-nulo; con el fin de elevar y mantener los niveles adecuados en plasma e intersticio pulmonar 11. El pronóstico de la DAAT es variable, y la mayoría de las personas con una deficiencia severa tienen menor esperanza de vida en relación con la población general, a excepción de los no fumadores que fueron identificados mediante cribado familiar o poblacional 6,12. El inicio precoz de la terapia aumentativa con AAT también incide en el pronóstico 13,14.
El objetivo de esta publicación es la descripción de un paciente atendido en una institución de salud en Barranquilla, Colombia, y explica la importancia de realizar un diagnóstico diferencial en pacientes con serio compromiso de la función pulmonar, que, a pesar de ser poco frecuente, debe vislumbrarse como posibilidad diagnóstica.
Metodología
Se describe el caso de un paciente masculino de 61 años con diagnóstico de (DAAT). Se llevó a cabo la recopilación de información para el análisis del caso clínico en fuentes de información como PubMed y Scopus, utilizando las palabras clave "déficit de alfa 1-antitripsina", "enfisema pulmonar", "enfermedad pulmonar obstructiva crónica" “EPOC de alfa 1-antitripsina".
Presentación del caso
Paciente masculino de 61 años, natural de San Juan de Nepomuceno, Bolívar y residente en Soledad, Atlántico (Colombia), quien fue derivado por medicina interna a la consulta de neumología. Presenta antecedentes de hipertensión arterial e hiperplasia prostática benigna, un episodio único de neumonía en 2018 e infección por SARS-CoV-2 leve sin secuelas reportadas. Experimenta un cuadro clínico de 3 años de evolución, caracterizado por disnea de moderados esfuerzos con un deterioro progresivo de su clase funcional, mMRC 3 (Escala de disnea modificada de la MRC), no asociado a otros síntomas. Previamente se descartó patología cardíaca como causa de la disnea. Niega antecedentes de tabaquismo o exposición pasiva a este, aunque refiere exposición a biomasa por corto tiempo en lugar abierto. Actualmente, en tratamiento médico con Losartán, Metoprolol, Amlodipino y Budesonida/formoterol 160/4,5 mcg 1 puff inhalado cada 12 horas.
En el examen físico, se observa un bajo peso según medidas antropométricas, cifras tensionales en 150/90 mmHg, sin taquicardia ni taquipnea, con adecuada saturación de oxígeno a FiO2 (fracción inspirada de oxígeno) ambiente. Al examen de tórax, se evidencia pectus excavatum leve, con aumento del diámetro anteroposterior del tórax, ruidos cardíacos rítmicos, sin evidencia de soplos y con ruidos respiratorios disminuidos de manera difusa. No presenta acropaquías ni otros hallazgos sugestivos de neumopatía crónica.
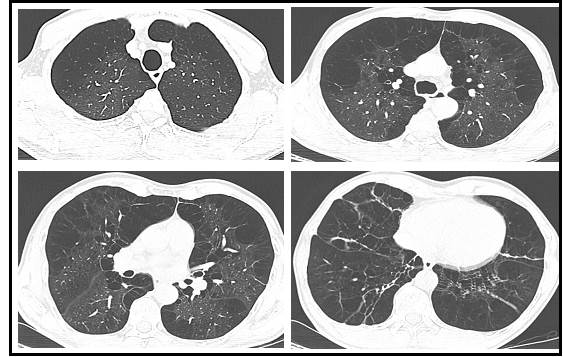
Aporta reportes de hemograma tipo IV con leucocitos 5.680/mm³, neutrófilos 53,3 %, linfocitos 29,1 %, eosinófilos 5,4 %, hemoglobina 16,8 g/dl, hematocrito 49,2 % y plaquetas de 246.000/mm³. La espirometría con broncodilatador muestra un índice de tiffeneau de 0,444, VEF1 (Volumen Espiratorio Forzado en un segundo): 58 % y la Capacidad Vital Forzada (CVF) del 106 % sin respuesta a broncodilatador. El ecocardiograma no revela alteraciones estructurales o de contractibilidad, con baja probabilidad de hipertensión pulmonar y una fracción de eyección preservada del 60 %. La tomografía de tórax sugiere hallazgos de enfisema centroacinar, paraseptal y paraacinar de predominio en bases, asociado a un aumento del volumen pulmonar (ver Figura 1).
Figura 1
Tomografía computarizada de tórax que muestra el patrón de enfisema centro acinar, paraseptal y panacinar con mayor compromiso en las zonas inferiores. Fuente: archivos del caso.
Se consideró descartar deficiencia de AAT, dada la ausencia de factores de riesgo exposicionales, en conjunto con los hallazgos tomográficos y espirométricos, además de la ampliación de pruebas de función pulmonar. Se ajustó el manejo inhalador con LABA/LAMA (LABA: beta-2 adrenérgicos de larga duración; LAMA: antimuscarínicos o anticolinérgicos de larga duración).
En el siguiente control, el paciente mostró una discreta mejoría sintomática. El informe de volúmenes pulmonares sugiere hiperinsuflación y alteración en DLCO corregido del 40%. Los niveles de AAT se encontraron en 26,5 mg/dl (valor de referencia: 90-200 mg/dl), y se realizó un estudio genotípico que informó la presencia de alelos PI*Z (PI*ZZ), considerándose una DAAT grave.
Se indicó continuar con la terapia inhalada y se formuló un inhibidor de alfa 1-proteinasa para infusión semanal, con una dosis de 60 mg/kg. Fue evaluado por gastroenterología, excluyendo compromiso hepático hasta la fecha de la valoración. La ecografía abdominal mostró un hígado de forma y tamaño normales, con eco-estructura conservada, sin alteraciones en las pruebas hepáticas y un informe de elastografía sin datos de fibrosis o esteatosis hepática. El tamizaje familiar a sus dos hijos demostró la presencia de PI*MZ y PI*ZI; lo que sugiere que la madre del segundo hijo es portadora, en principio, de un alelo PI*I.
Actualmente, el paciente continúa recibiendo terapia con inhibidor de alfa 1-proteinasa para infusión semanal y está bajo seguimiento estricto por el servicio de neumología.
Discusión
La DAAT es un trastorno hereditario poco reconocido clínicamente que afecta los pulmones, el hígado y, en raras ocasiones, la piel. No obstante, un análisis sanguíneo de los niveles AAT con confirmación mediante genotipo permite en la mayoría de los casos identificar fácilmente el DAAT 15, el diagnóstico inicial puede ser complejo debido a su baja sospecha, entre cinco a diez años, la baja pesquisa de niveles de AAT, la superposición con el consumo de tabaco y otros factores de exposición, así como la probabilidad de una presentación clínica gradual similar 16, generando un retraso diagnóstico 17,18.
Se calcula que entre 180.000 y 1,2 millones de personas en todo el mundo presentan los fenotipos PI*ZZ y PI*SZ, respectivamente. Sin embargo, alrededor del 90 % de estos casos aún no han sido diagnosticados. En Brasil, se ha registrado una prevalencia de 38,0 casos por cada 1.000 habitantes para el fenotipo PI*SZ y de 6,5 casos por cada 1.000 habitantes para el fenotipo PI*ZZ 4.
La variante Z de AAT suele tener niveles de AAT circulantes entre el 10 % y el 15 % del valor normal y se caracteriza por la sustitución de un aminoácido de lisina por ácido glutámico en la posición 342 18. La presencia de dos copias del alelo Z mutante se asocia con DAAT severa, Además, se ha sugerido un umbral sérico mínimo de 57 mg/dl (11 µmol/L) como un punto crítico en el cual la cantidad de AAT es insuficiente para inhibir la elastasa de neutrófilos, generando deficiencia en los procesos de protección del tejido pulmonar. Sin embargo, es importante señalar que esta propuesta de nivel sérico carece de evidencia científica sólida 19 debido a las limitaciones para su verificación precisa. Además, la mayoría de los individuos que se encuentran por debajo de estos niveles corresponden a genotipos como PI*SZ, PI*ZZ y homocigotos para variantes raras o nulas, quienes son propensos a la destrucción del tejido pulmonar durante los procesos inflamatorios, con el posterior desarrollo de enfisema panacinar 20,21.
Se ha descrito una diferencia sustancial en el fenotipo pulmonar y el pronóstico entre los pacientes con genotipo de proteínas PI*SZ y los que son PI*ZZ. Los sujetos PI*SZ son menos susceptibles al humo del cigarrillo que los individuos PI*ZZ. Se ha observado que la proporción de casos de enfisema y EPOC en pacientes PI*ZZ ocurren muy frecuentemente con un índice tabáquico menor de 20 paquetes-año 18. Esto sugiere que incluso una cantidad relativamente baja de exposición al tabaco puede tener un impacto significativo en la función pulmonar de los pacientes con el genotipo PI*ZZ 22, resaltando la importancia de considerar la susceptibilidad al cigarrillo al evaluar el impacto de la DAAT en los pacientes con diferentes genotipos.
Dentro los hallazgos reportados por tomografía, también se han descrito divergencia en las anormalidades enfisematosas entre individuos homocigotos PI*ZZ y PI*SZ, siendo más prominente el compromiso basal y en los vértices en el primero, 64% y 36 % respectivamente. A diferencia los individuos PI*SZ, 42,9 % tienen enfisema dominante en la zona superior 23,24.
Los pacientes con DAAT pueden presentar limitación grave del flujo de aire que a menudo es desproporcionada con respecto a su historial de tabaquismo y el grado de obstrucción de las vías respiratorias. Frecuentemente, la limitación del flujo de aire no es fija y, en consecuencia, existe una amplia variación en la respuesta broncodilatadora 16,18. La presentación más frecuente de obstrucción del flujo aéreo en pacientes con DAAT es a través de la prueba de espirometría, donde se observa un patrón obstructivo con una relación VEF1/CVF inferior al 70 %. En los pacientes con asma y DAAT, los resultados positivos de la prueba broncodilatadora, sugieren la presencia de hiperreactividad bronquial y la necesidad de evaluar y tratar adecuadamente esta condición respiratoria 25,26.
El paciente del caso se encontraba en una fase avanzada de la enfermedad en el momento del contacto inicial, con un compromiso enfisematoso extenso. Se destaca la evidencia de una obstrucción fija del flujo aéreo sin respuesta al broncodilatador, una capacidad de difusión de CO2 disminuida y volúmenes pulmonares que demostraban hiperinsuflación. Estos hallazgos se correlacionan con el grado de disnea y el compromiso de la calidad de vida del paciente. Su origen socioeconómico y cultural bajo pudo favorecer una baja posibilidad de consulta temprana, además de la baja sospecha diagnóstica diferencial en los niveles menores de atención, considerando la ausencia de compromiso exposicional de importancia. La identificación principalmente de pacientes con genotipo PI*Z (PI*ZZ) van de la mano con el fenotipo presentado por el paciente, DAAT grave, aunque también en pacientes con genotipos PI*SZ, variantes raras y nulas en estado de homocigosis.
Se ha descrito en reportes clínicos previos una presentación clínica y paraclínica similar al paciente reportado, evidenciando un compromiso grave de la función y arquitectura pulmonar al momento del diagnóstico. Esto suele estar relacionado con el grupo etario mayor de 50 años, la ausencia de factor exposicional y el genotipo PI*Z (PI*ZZ) 27.
El manejo de la DAAT varía según la presentación de la enfermedad. Aunque en la actualidad, existen fármacos para el manejo de la enfermedad hepática relacionada, estos se encuentran aún en estudio 28, por lo que la terapia aumentativa es el único tratamiento aprobado para el tratar la enfermedad pulmonar relacionada 29,30.
La terapia aumentativa consiste en la infusión intravenosa de AAT purificada proveniente de sangre de donantes humanos sanos, ha demostrado ser segura y bien tolerada 31; aumentando la concentración de AAT plasmática en pacientes con EPOC asociado a DAAT grave, no fumadores o exfumadores, con VEF1 entre 30 a 65% y con pérdida de la función pulmonar o progresión del enfisema. Las dosis habituales son de 60 mg/kg una vez por semana 6,17. Estudios observacionales relacionados con el análisis de la eficacia clínica de la terapia aumentativa evidencian un descenso en la pérdida de la función pulmonar, determinada por el descenso del VEF1 y una reducción en la presencia de infecciones en los pulmones; además, prolonga el tiempo de supervivencia y reduce la necesidad de trasplante de pulmón o la invalidez y la mortalidad 30,32,33,34. Datos emergentes muestran una probable utilidad en pacientes que no cumplen con los criterios previamente establecidos 35. Por otro lado, hay datos emergentes que muestran una probable utilidad en pacientes que no cumplen con los criterios previamente establecidos 35.
Conclusión
Los pacientes con sintomatología respiratoria, compromiso pulmonar importante en pruebas de función pulmonar y hallazgos imagenológicos compatibles con enfisema pulmonar de predominio en bases y/o panlobar, deben ser enmarcados como fuertemente sospechosos de DAAT, inclusive si hay ausencia o pocos factores de riesgo.
En todo paciente con EPOC, asma y otras patologías respiratorias se sugiere realizar un tamizaje de los niveles AAT por lo menos una vez en la vida, que permita diagnosticar oportunamente e iniciar de forma temprana, un tratamiento de renovación en pacientes con indicación de uso; impactando así la calidad de vida y pronóstico de los pacientes afectados.